
Décès d’enfants causés par la codéine, une conséquence du métabolisme
Le métabolisme des opioïdes, avec son cortège de « métabolites actifs », ou autres « cytochromes » est d’un abord souvent hermétique pour les cliniciens. Le discours des firmes prête lui aussi à confusion, tentant d’utiliser des notions de pharmacologie comme autant d’arguments marketing en faveur de leurs médicaments. Pourtant, ce paramètre pharmacologique est moins négligeable qu’il n’y paraît et peut influencer de façon notable la sécurité et l’efficacité de molécules que nous employons quotidiennement.
Recommandations
L’actualité dans la douleur a été marquée par les restrictions récentes imposées à la codéine : « contre-indication chez la femme allaitante, chez le patient “métaboliseurs rapides” ou encore chez l’enfant de moins de 12 ans ». Ces interdictions ont été formulées par les autorités sanitaires européennes et françaises sur la base de cas « graves, voire mortels, de dépression respiratoire » rapportés outre-Atlantique. Leur origine se retrouve dans le métabolisme : les « métaboliseurs rapides », convertissant de façon disproportionnée la codéine en son métabolite morphine, se sont retrouvés surdosés [1].
Début 2016, la Haute Autorité de santé (HAS), appuyée par un comité de professionnels de santé, a formulé de nouvelles recommandations pour la prise en charge de la douleur chez l’enfant. Ces dernières écartent l’utilisation de la codéine et alertent sur celle du tramadol, dont le métabolisme s’effectue aussi par le CYP450 2D6 (nous y reviendrons). En alternative, la HAS préconise en première intention le paracétamol et l’ibuprofène. La morphine est « la molécule de choix » quand les douleurs sont intenses [2].
Plus récemment, l’American Academy of Pediatrics va dans le même sens avec son rapport « Codein: time to say no » : la codéine et les autres opioïdes qui empruntent les cytochromes exposent à des effets fortement variables d’un individu à l’autre, pouvant provoquer des effets imprévisibles [3]. S’il est toujours judicieux de se préoccuper de la sécurité des médicaments chez les enfants, il est nécessaire de rappeler que les variations du métabolisme touchent également les adultes. Les sujets âgés sont aussi vulnérables que les enfants. Une métabolisation importante de la codéine et du tramadol peut entraîner des conséquences dramatiques en termes de surdosage (ou de non soulagement de la douleur). Les patients en gériatrie reçoivent de surcroît de nombreux médicaments susceptibles d’altérer le métabolisme des opioïdes empruntant le CYP450 2D6 (antidépresseurs, antifongiques, médicaments pour les troubles cardiovasculaires) [4].
En septembre 2016, nous avons livré une première réflexion sur le sujet par l’intermédiaire d’une newsletter diffusée aux correspondants de la revue Le Flyer : « Analgésiques morphiniques et 2D6, attention à la codéine et à d’autres opioïdes » [5].
Cet article en est le prolongement.
• Dans un premier temps, nous reviendrons sur quelques généralités sur le métabolisme des médicaments.
• Puis dans un second temps, nous nous intéresserons spécifiquement au métabolisme des opioïdes, à ses conséquences et nous proposerons quelques règles pour minimiser les risques.
Les médicaments sous influence des spécificités du métabolisme [6, 7]
Après son administration, un médicament va passer par différentes étapes qui vont influencer son devenir. L’acronyme ADME, cher aux pharmacologues, liste ces quatre étapes : absorption, distribution, métabolisme et élimination.
Le métabolisme est une de ces étapes clés. Elle s’effectue principalement dans le foie sous l’action d’enzymes :
• les cytochromes, dits CYP, responsables du métabolisme de 75 % des traitements existants. Il s’agit d’une famille composée de très nombreuses enzymes (1A2, 3A4, 2D6, 2E1…) ;
• et les autres enzymes, au nom un peu barbare (UGT : uridine 5’-diphosphoglucuronyltransférase…).
Les cytochromes
Selon le CYP considéré, la quantité comme la fonctionnalité peuvent être très variables d’une personne à l’autre, ou pour une même personne au cours du temps :
• les gènes codant pour certains CYP présentent des variations. Le CYP 2D6 est le plus concerné par cette variabilité. Or il est responsable de la métabolisation de la plupart des opioïdes (hormis la morphine et l’hydromorphone) ; un individu porteur d’un gène altéré dont le code aboutit à la synthèse d’un CYP 2D6 peu ou non fonctionnel sera dit métaboliseur lent ou intermédiaire. Inversement, un individu peut être porteur de nombreuses copies du gène, aboutissant à une quantité plus importante de CYP 2D6, il sera métaboliseur ultra-rapide. Les individus ne possédant que deux copies du gène fonctionnel seront qualifiés de métaboliseurs extensifs avec des conséquences variables sur un plan clinique et selon les molécules ;
• les CYP sont à l’origine de nombreuses interactions médicamenteuses. Certaines substances dites « inducteurs enzymatiques » vont entraîner une synthèse accrue de CYP et donc une accélération de leur activité. À l’inverse, les « inhibiteurs enzymatiques » vont perturber le fonctionnement de l’enzyme et donc la ralentir. De nombreux médicaments sont susceptibles d’interagir avec les CYP et, par suite, de modifier le métabolisme des opioïdes.
Opioïdes et cytochromes
Les opioïdes transformés par les CYP, en exposant à des effets imprévisibles, nécessitent une attention toute particulière. Prenons l’exemple de la codéine, elle est principalement métabolisée par le CYP 3A4 en un métabolite inactif et une petite partie est transformée par le CYP 2D6 en morphine. C’est cette fraction qui est principalement responsable de l’activité antalgique. Un patient métaboliseur ultra-rapide du 2D6 présentera une répartition différente, avec une plus forte proportion de morphine, ce qui l’exposera davantage à des effets opiacés, pouvant même aboutir à une situation de surdosage. Il sera aussi plus vulnérable aux interactions médicamenteuses, à l’origine d’une toxicité potentiellement plus prononcée.
En pratique, lorsque la prescription d’un opioïde est envisagée, deux interrogations peuvent aider à en anticiper les effets :
• Par quelles enzymes l’opioïde est-il métabolisé : CYP ou autre ?
• L’opioïde donne-t-il des métabolites actifs, susceptibles de s’accumuler ?
Les conséquences cliniques et les règles de précaution à adopter seront variables d’un opioïde à l’autre.
Une attention particulière à porter aux opioïdes métabolisés par les cytochromes
De façon schématique, les opioïdes les plus couramment utilisés peuvent être divisés en trois groupes (Tab. 1) :
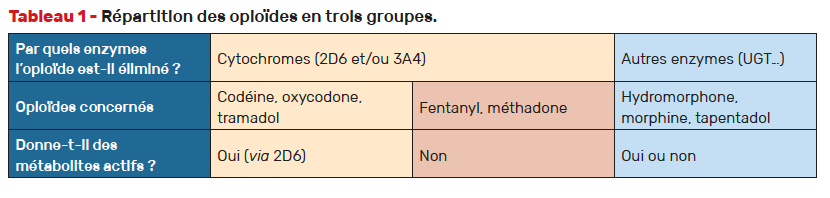
1er groupe : codéine, oxycodone et tramadol, attention aux métabolites actifs
Codéine, tramadol et oxycodone empruntent tous trois les cytochromes (2D6 et 3A4). Le passage par le 2D6 donne des métabolites actifs [6-8] :
• la codéine, inactive en elle-même, est métabolisée en morphine ;
• l’oxycodone se transforme en oxymorphone (métabolite 14 fois plus actif que l’oxycodone)
• et le tramadol donne le métabolite actif M1.
Métaboliseurs ultra-rapides
On estime qu’environ 5 à 10 % de la population seraient des métaboliseurs ultra-rapides du 2D6 (les chiffres variant selon les populations). Ces personnes, en transformant de manière plus prononcée la molécule en son métabolite actif, pourront être exposées à posologie usuelle à une toxicité où à un risque de surdosage [9].
Métaboliseurs lents ou intermédiaires
À l’inverse, entre 15 et 25 % des patients seraient des métaboliseurs lents ou intermédiaires. Ils nécessiteront une posologie plus importante pour ressentir les effets du traitement ou ne ressentiront pas les effets antalgiques [9].
Pour près d’un tiers des patients, les effets de ces opioïdes seront donc imprévisibles :
• les décès d’enfants survenus après administration de codéine sont survenus chez des métaboliseurs rapides [1, 2] ;
• dans une étude publiée en 2010, l’équipe suisse du Dr Jules Desmeules s’est penchée sur l’oxycodone et concluait que « son profil métabolique jouait un rôle majeur sur ses effets analgésiques et sa toxicité » [10, 11].
Le polymorphisme génétique n’est pas le seul paramètre influençant les effets antalgiques des opioïdes. La métabolisation par les cytochromes entraîne aussi un risque accru d’interactions médicamenteuses [6].
Certaines molécules peuvent diminuer l’activité des cytochromes (inhibiteurs enzymatiques) :
• s’il s’agit d’un inhibiteur du CYP 2D6, son association à la codéine, à l’oxycodone ou au tramadol limitera la production de métabolites actifs et l’efficacité antalgique. On retrouve dans ce cas certains antidépresseurs (paroxétine, fluoxétine, duloxétine) ou des antifongiques (terbinafine) ;
• à l’inverse, l’association d’un inhibiteur du CYP 3A4 favorisera la voie métabolique passant par le CYP 2D6 et accélérera donc la production de métabolites actifs. L’association à certains antifongiques (kétoconazole), antibiotiques (érythromycine, clarithromycine…) ou molécules à visée cardiovasculaire (amiodarone, diltiazem) qui sont connus pour inhiber le CYP 3A4 augmentera le risque de surdosage.
Ce dernier mécanisme a fait l’objet d’une alerte pour l’oxycodone aux États-Unis. Les autorités sanitaires américaines (FDA) ont apposé un « black-box warning », plus haut niveau d’avertissement pour un médicament, informant du risque de dépression respiratoire potentiellement fatal en cas d’association à un inhibiteur du 3A4 [6].
2e groupe : fentanyl et méthadone, des métabolites inactifs via le 3A4
Fentanyl et méthadone donnent des métabolites inactifs essentiellement par le CYP 3A4. Leur efficacité antalgique repose sur la molécule mère. Elle peut être influencée par le polymorphisme génétique, mais dans une moindre proportion par rapport aux opioïdes empruntant le 2D6 (codéine, tramadol et oxycodone) [7, 8].
Si le profil pharmacologique et l’absence de métabolites actifs pour le fentanyl et la méthadone les rendent intéressants dans certaines situations (insuffisance rénale…), ils sont sujets à de nombreuses interactions médicamenteuses [6] :
• l’association d’un inhibiteur du 3A4 pourra entraîner des surdosages ;
• à l’inverse, un inducteur enzymatique du 3A4 provoquera une diminution de l’efficacité antalgique. La rifampicine est l’exemple cité comme le plus spectaculaire [12]. Carbamazépine et phénytoïne sont d’autres molécules probablement plus fréquentes en pratique courante.
Pour la méthadone, d’autres cytochromes sont impliqués. Dans un objectif de simplification, nous nous sommes concentrés sur le 3A4. L’article « Méthadone et interactions médicamenteuses » des Drs Maroussia Wilquin et Stéphane Robinet revient plus en détails sur cette molécule [13].
3e groupe : hydromorphone, morphine, tapentadol, pas de passage par les cytochromes
Le métabolisme de l’hydromorphone, de la morphine et du tapentadol ne passe pas par les cytochromes mais par enzymes de phase II (exemple : UGT) [7, 8]. Ils peuvent entraîner la formation de métabolites actifs, amenant en cas d’insuffisance rénale marquée (DFG < 60 ml/min) à des règles de précautions identiques à celles de la codéine, de l’oxycodone ou du tramadol [14].
La métabolisation de ces molécules les rend cependant intéressantes pour d’autres raisons :
• elles sont très peu sujettes au polymorphisme génétique, les quelques publications s’y étant intéressées ne retrouvant pas de conséquence clinique significative ;
• elles exposent moins aux interactions pharmacocinétiques : l’hydromorphone n’en a pas et la morphine en a peu (rifampicine).
Ces particularités pharmacologiques ont conduit certains auteurs à privilégier l’hydromorphone et la morphine en cas d’altération de la fonction hépatique, en pédiatrie ou pour des situations nécessitant des polyprescriptions (gériatrie, oncologie…). En pratique, le choix de ces molécules permet de s’affranchir des effets inattendus liés au polymorphisme génétique ou aux interactions pharmacocinétiques [8].
Quelles sont les précautions à adopter ?
Chaque situation sera à évaluer au cas par cas. Lorsque la prescription d’un opioïde est envisagée, il faudra s’assurer au préalable d’en maîtriser la pharmacologie :
• Comment l’opioïde est-il métabolisé ?
• Donne-t-il des métabolites actifs ?
• Existe-t-il un risque d’interaction médicamenteuse ?
• D’autres éléments sont-ils à prendre en considération (insuffisance hépatique, rénale…) ?
L’instauration du traitement et son adaptation seront fondées sur l’évaluation clinique. En complément, quelques règles d’ordre général peuvent être formulées.
Pour éviter des effets imprévisibles, privilégier les opioïdes n’empruntant pas les CYP [8]
En l’absence de test génétique de routine, il n’est pas possible de savoir si un patient est métaboliseur lent ou ultra-rapide du CYP 2D6. De même, la pleine maîtrise des co-prescriptions responsables d’interactions médicamenteuses n’est pas non plus toujours faisable. L’utilisation de molécules éliminées par les CYP 2D6 et 3A4 expose donc à la survenue d’effets imprévisibles.
La poudre d’opium semblerait être une alternative à la codéine et au tramadol. Cependant, si son métabolisme n’empruntait pas les CYP, elle présenterait, de par sa composition inconstante, des risques semblables à la morphine pour une efficacité imprévisible [15].
Les opioïdes forts ayant tous une efficacité équivalente, il semble plus judicieux de privilégier les molécules qui n’empruntent pas les cytochromes lorsque cela est possible. L’utilisation de morphine, hydromorphone ou tapentadol minimise les incertitudes liées au polymorphisme et aux interactions.
L’oxycodone est une molécule qui a sa place dans la prise en charge de la douleur mais, en lien avec les variations métaboliques qu’elle subit, elle semble être une alternative de seconde intention.
Évaluer constamment les co-prescriptions [16]
Si la prescription d’un opioïde sujet aux interactions pharmacocinétiques est envisagée, il sera nécessaire de renforcer la surveillance clinique à l’instauration du traitement, tout au long de son utilisation, ainsi qu’à son arrêt. La posologie présentant la meilleure balance efficacité/sécurité sera à questionner régulièrement. Elle est susceptible de varier de façon importante au cours du temps ou au gré des interactions médicamenteuses : changement de traitements concomitants, ajout d’une nouvelle thérapeutique…
Se méfier des ratios d’équianalgésie [17]
De multiples publications proposent des ratios de conversion en cas de changement d’un opioïde à l’autre, avec des variations selon les sources. Plusieurs auteurs [17] alertent sur leur utilisation stricto sensu : ces ratios ont pour la plupart été déterminés dans des études en dose unique ou avec de petits effectifs. Ils doivent être utilisés à titre indicatif tant les facteurs de variabilité sont nombreux : sens de changement de l’opioïde, posologie de départ, variabilité interindividuelles…
Si le polymorphisme génétique et les interactions médicamenteuses via les cytochromes ne sont pas les seuls paramètres à considérer, ils constituent une inconnue supplémentaire. En cas de changement, il apparaît hasardeux de se limiter à la stricte application des ratios d’équianalgésie. Le principe de titration doit rester une règle en cas de changement d’opioïde avec des ratios de conversion prudents.
Travailler en réseau pour surveiller les interactions
Pour de nombreuses pathologies ou situations cliniques, il n’est pas rare que les prescripteurs se succèdent. Le risque de polymédication augmente avec la multiplication des intervenants et la reconduction systématique (“copier/coller”) des anciennes prescriptions qui s’accumulent tout au long du périple du patient. Dans la mesure du possible, il est préférable qu’un seul et même prescripteur centralise les prescriptions (que ce soit en établissement de santé ou en médecine libérale).
La surveillance clinique apporte souvent des informations précieuses quant à l’efficacité des traitements et l’existence d’interactions médicamenteuses : apparition soudaine et inattendue d’une somnolence ? Résurgence de douleurs en l’absence d’évolution de la pathologie ?
Les équipes soignantes (médecins, infirmier(e)s) ainsi que les pharmaciens, tiennent une place de premier plan dans le suivi du traitement opioïdes et dans sa surveillance.
Conclusion
Choisir un opioïde analgésique doit relever d’éléments reposant sur une pratique clinique, elle-même influencée par de solides connaissances pharmacologiques. Il ne faut pas oublier que les arguments marketing des laboratoires sont toujours très bien faits et séduisants. La simple influence des firmes pharmaceutiques qui vantent les mérites de fausses nouveautés sur la base d’allégations plutôt que de publications doit être écartée au moment du choix de prescrire un opioïde analgésique plutôt qu’un autre [18]. Le slogan « plus efficace et mieux toléré » résiste rarement à l’examen des faits et de la littérature scientifique. Dans les faits, la morphine reste le traitement opioïde fort de première intention. L’Esmo, par exemple, va dans ce sens [19]. D’autres sociétés savantes, comme l’European Association for Palliative Care, ne se prononcent pas sur un opioïde de première intention plutôt qu’un autre. Le choix peut donc se faire sur sa maniabilité, l’existence d’une large gamme de dosages et formes galéniques. Le prix, à efficacité comparable, doit également intervenir comme critère de choix.
Dans certains cas, la méthadone présente un profil intéressant, notamment en cas d’insuffisance rénale, et parce qu’elle est aussi un antagoniste des récepteurs NMDA, mais sa maniabilité délicate en fait plutôt un traitement de seconde intention. Le fentanyl propose une voie d’administration différente (les patches) et un métabolisme intéressant en cas d’insuffisance rénale.
Les formes transmuqueuses, compte tenu du risque addictif qu’elles présentent, doivent être strictement réservées aux accès douloureux paroxystiques (ADP).
Les paliers 2, très utilisés en France, sont très concernés par les variations métaboliques. S’ils restent une option en cas de douleurs modérées, leur efficacité et leur tolérance est moins bonne que la morphine à faible dose, si l’on se réfère à la récente publication de Bandieri et al. [20].
Les auteurs déclarent ne pas avoir de lien d’intérêt.
Bibliographie
1. HAS. Codéine : Avis de la commission de la transparence [en ligne]. In HAS [consulté le 02/03/23]. Septembre – octobre 2016. Disponible sur : www.has-sante.fr/upload/docs/evamed/CT-13350_PARACETAMOL_CODEINE_TEVA_PIS_RI_Avis2_CT13350.pdf.
2. HAS. Prise en charge médicamenteuse de la douleur chez l’enfant : alternatives à la codéine. Fiche memo [en ligne]. In HAS [consulté le 02/03/23]. Janvier 2016. Disponible sur : www.has-sante.fr/jcms/c_2010340/fr/prise-en-charge-medicamenteuse-de-la-douleur-chez-l-enfant-alternatives-a-la-codeine.
3. Tobias JD, Green TP, Coté CJ. AAP section on anesthesiology and pain medicine, aap committee on drugs. Codeine: Time To Say “No”. Pediatrics 2016 ; 138 : e20162396.
4. Lynch T. Management of drug-drug interactions: considerations for Special Populations—A focus on Opioid use in the elderly and Long Term care. Am J Manag Care 2011 ; 17 : S293-8.
5. Annequin D, Kieffert P, Pouymayou J, Robinet S. Newsletter : analgésiques morphiniques et 2D6, attention à la codéine et à d’autres opioïdes. Septembre 2016.
6. Overholser BR, Foster DR. Opioid pharmacokinetic Drug-Drug Interactions. Am J Manag Care 2011 ; 17 : S276-87.
7. Smith H. Opioid Metabolism. Mayo Clin Proc 2009 ; 84 : 613-24.
8. Mercadante S. Opioid metabolism and clinical aspects. European Journal of Pharmacology 2015 ; 769 : 71–8.
9. Meyer UA. Pharmacogenetics – five decades of therapeutic lessons from genetic diversity. Nature Reviews Genetics 2004 ; 5 : 669-76.
10. Samer CF, Daali Y, Wagner M et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. British Journal of Pharmacology 2010 ; 160 : 919–30.
11. Samer CF, Daali Y, Wagner M et al. The effects of CYP 2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. British Journal of Pharmacology 2010 ; 160 : 907–18.
12. Niemi M, Backman JT, Fromm MF et al. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 2003 ; 42 : 819-50.
13. Wilquin M, Robinet S. Interactions médicamenteuses avec la méthadone et implications pratiques. Flyer 57. Novembre 2014.
14. Sande TA, Laird BJA, Fallon MT. The use of opioids in cancer patients with renal impairment—a systematic review. Support Care Cancer 2017 ; 25 : 661-75.
15. Prescrire. « Izalgi° : dosage élevé d’une association paracétamol + opium pour la voie orale ». Rev prescrire 2015 ; 35 : 821.
16. Fanciullo GJ, Washington T. Best Practices to reduce the risk of drug-drug interactions: Opportunities for managed care. Am J Manag Care 2011 ; 17 : S299-304.
17. Roulet L, Escher Imhop M, Piguet V et al. Rotation des opioïdes : de la théorie à la pratique. Recommandations interdisciplinaires du réseau douleur des HUG. Rev Med Suisse 2011 ; 7 : 1400-6.
18. Poumayou J et Robinet S. Oxycodone, objet marketing ou alternative à la morphine. Flyer 63, mai 2016.
19. Ripamonti CI, Santini D, Maranzano E et al. On behalf of the ESMO Guidelines Working Group; Management of cancer pain: ESMO Clinical Practice Guidelines. Ann Oncol 2012 ; 23 : vii139-54.
20. Bandieri E, Romero M, Ripamonti CI et al. Randomized trial of low-dose morphine versus weak opioids in moderate cancer pain. Journal of Clinical Oncology 2016 ; 34 : 436-42.

